miRNA annotation: complex scenarios
Everybody who is working with microRNA knows about miRBase, it was the first miRNA catalogue. Everybody is using it to annotate small RNA sequences as miRNA or not. And it is great, and very helpfully…but there are some cases that we should investigate our results.
For instance, what happens with miR-1246? it is a recent miRNA, primate specific, detected by sequencing in different studies (1,2,3). The counts related to these sequences are not so high, actually very low after normalization, but still are there. miRBase gives a precursor this region: chr2: 177465708-177465780 [-]

We see that there is a repeat element (ERV, retrovirus) and also it is a pretty conserved region. Until here, everything allright. But, I realized some time ago, annotating some data that I had, that the mature miRNA annotates in other regions:
| chr:start-end strand | type | conservation |
| chr13:21,186,304-21,186,473 + | U2 | human |
| chr14:96,850,961-96,851,148 + | U2 | rhesus |
| chr5:157,403,780-157,403,964 - | U2 | rhesus |
| chr6:89,773,226-89,773,409 - | U2 | rhesus |
| chr13:46,948,540-46,948,725 - | U2 | rhesus |
| chr3:127,793,920-127,794,092 - | U2 | rhesus |
| chr2:177,465,708-177,465,780 - | miR-1246 | rhesus,mouse,dog,elephant |
All of them are copies of a RNA repeat according RepBase, specifically the snRNA U2 class (150-200 nt). In addition, I got much more reads inside those (extra) locations than inside the annotated precursor (3 orders bigger). Furthermore, all of them had a peak in their coverage profile (fig 2).

The question that arises now: is this a real miRNA, or a sub-product, or both? with any function? Maybe there is more than one precursor, or maybe it is a miRNA-like snRNA. Moreover, the secondary structure of these U2 is very similar to a miRNA precursor hairpin (fig 3).
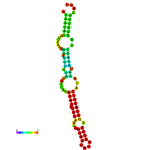
One cause of this annotation could be that the unique precursor conserved beyond primates is the one mentioned in miRBase, while the other U2 locations are primate-specific. Extra analyses should be done to reveal the real nature of this miRNA. Results suggest that, either the miRNA has more than one precursor, or a sub-product, or both.
As an extra-analysis, I took public data, KO Dicer and control, of HELA cells (GSM769512) to study the dependency od these sequences to DICER. You expect a decrease in the number of counts of miRNA sequences, between control and KO dicer. The exact miRNA sequence decrease 1/3 in KO dicer cells, assuming a Dicer dependency, and take this sequence as a real miRNA. But, we have a lot of other sequences in the U2 regions, but not in the annotated precursor, and only differ in one extra nucleotide (Adenosine) at the beginning (miR-1246: AAUGGAUUUUUGGAGCAGG and U2: AAAUGGAUUUUUGGAGCAGG).This sequence can be annotated as miR-1246 if you allow 1 mismatch in the alignment (the A nt). This sequence had the same counts in normal cells and KO cells, suggesting that sequences from U2 are not Dicer-dependent. Summarizing, we have the mature annotated miRNA mapping in all these positions, and in addition, we have other sequences (one nt longer), that only map in the U2 positions. So, how we can differentiate what proportion of sequences comes from the precursors and what comes from the other positions? Well, one possibility is to detect the precursor by PCR in your samples, to assume that at least it is there, or try to see whether the coverage in the different locations makes sense more with one scenario or another…Or maybe it is not a big deal, you have this small RNA, the question is whether it has any regulatory function or not.
From here, we may agree that it is better to double-check what is happening when annotating small RNAs, because there are many possibilities to take a wrong conclusion if you only believe in a preliminary analysis. Nowadays, data is increasing a lot, and scientists need to make a bunch of assumptions to obtain something, but be careful with those assumptions (indeed, they are needed to evolve in science) because you may be missing something else or getting something wrong or wasting your time with experiments to corroborate your computational results.
References
[1] PMID:18583537
“MicroRNA discovery and profiling in human embryonic stem cells by deep sequencing of small RNA libraries”
Bar M, Wyman SK, Fritz BR, Qi J, Garg KS, Parkin RK, Kroh EM, Bendoraite A, Mitchell PS, Nelson AM, Ruzzo WL, Ware C, Radich JP, Gentleman R, Ruohola-Baker H, Tewari M
Stem Cells. 26:2496-2505(2008).
[2] PMID:20300190
“Characterization of the Melanoma miRNAome by Deep Sequencing”
Stark MS, Tyagi S, Nancarrow DJ, Boyle GM, Cook AL, Whiteman DC, Parsons PG, Schmidt C, Sturm RA, Hayward NK
PLoS One. 5:e9685(2010).
[3] PMID:20459774
“Ultra-high throughput sequencing-based small RNA discovery and discrete statistical biomarker analysis in a collection of cervical tumours and matched controls”
Witten D, Tibshirani R, Gu SG, Fire A, Lui WO
BMC Biol. 8:58(2010).