Multi-Lab Genomics Data Sharing with IGV, S3, and GitHub Actions
Composing existing tools to solve collaborative research infrastructure without a dev team
Most collaborative genomics projects fail not because the science is hard, but because managing and sharing heterogeneous data across collaborators is hard.
We solved this problem by composing existing tools instead of building custom code. Here’s how.
TLDR:
Thanks to Ruitong Li for contributing to the project.
The Problem
Your collaborators send differential expression results from their single-cell RNA and ATAC-seq experiments. Some work is in mouse, some in human. Your task: find the genes they identified as significant, check the signal in all other experiments, and compare across species—all from a single interface.
Without infrastructure, this means emailing files, manually looking up gene coordinates, translating between genome builds (mouse to human), and checking signal experiment by experiment. It doesn’t scale. Collaborators wait. Nobody wins.
The Solution: Composition Over Complexity
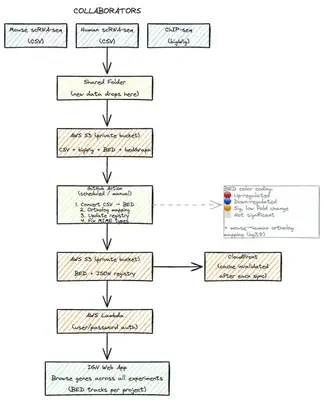
We focused on three decisions:
1. Choose Tools That Do 80% of the Work
We selected IGV (Integrative Genomics Viewer)—the web app version. It’s a professional genome browser designed for exactly what we needed: jumping between genes, loading multiple data types, fast interactive navigation. We didn’t build visualization code; IGV handles that elegantly.
2. Standardize Everything Into Compatible Formats
IGV natively understands a few core formats: BED files (genomic regions with annotations), BigWig files (signal tracks), and narrowPeak files (ATAC-seq peaks). Our collaborators send data in CSV format from DESeq2.
We built an automated pipeline that:
- Detects file types: ATAC-seq BigWig/BED files, RNA-seq CSV files
- Converts DESeq2 results → color-coded BED files with four-tier significance coloring:
- Red: Up-regulated (p < 0.05 & log2FC > 1.5)
- Blue: Down-regulated (p < 0.05 & log2FC < -1.5)
- Orange: Significant but low fold change (p < 0.05 & |log2FC| < 1.5)
- Gray: Not significant
- Translates cross-species data: If the DESeq2 results are from mouse, the pipeline maps mouse genes to human orthologs and converts coordinates to hg38—automatically
- Leaves compatible formats as-is: ATAC-seq peaks and BigWig files pass through unchanged
The script scans project folders, identifies each file type, applies the appropriate transformation, and generates a track registry describing everything for IGV.
3. Automate Everything, Including Deployment
We organized data hierarchically: three main research projects, with experiments nested inside. When you open IGV, you see a project modal. Click a project, and you get a searchable table of all its experiments—grouped by experiment ID or data type.
Everything lives on AWS S3. Lambda@Edge functions enforce password authentication at the CloudFront edge—only authorized collaborators can access the data. Passwords are shared securely with the team.
For continuous updates, we use GitHub Actions. Set it to run daily or trigger manually. When new data arrives in the S3 data folder, the action:
- Scans all projects for new CSV files
- Runs the conversion script on anything new (skips already-converted files)
- Updates the track registry (CSV and JSON files)
- Syncs metadata back to S3
- Fixes MIME types for BigWig files
- Invalidates the CloudFront cache
Within seconds, new data is live in IGV. No manual deployment. No command-line expertise required. The whole team benefits from new data immediately.
Technical Implementation
Here’s what makes this work technically:
Data Organization:
Three projects (project1, project2, project3), each containing experiment subfolders with BED, BigWig, or CSV files.
Automation Script (automate_igv_tracks.py):
- Auto-scans the entire data directory recursively
- Identifies ATAC-seq files (.bed, .narrowPeak, .bw) and RNA-seq CSV files
- Converts new CSV files to BED using the
create_igv_bed.pyconverter (skips if BED already exists) - Generates
project_name.csvwith track metadata (type, URL, display options) - Generates
project_name.jsonwith IGV configuration - Updates the central
trackRegistry.json - Optional:
--dry-runflag to preview changes before applying
Format Conversion (create_igv_bed.py):
- Reads DESeq2 output (gene_id, log2FoldChange, p-value columns)
- Species detection: Automatically recognizes mouse or human (looks for MGI gene symbols or Ensembl IDs)
- Species translation: Maps mouse genes to human orthologs and converts coordinates
- Creates color-coded BED files with biological meaning (red=up, blue=down, orange=borderline)
Deployment to Production:
- S3 bucket stores all data and configuration files
- CloudFront CDN sits in front for fast global access
- Lambda@Edge enforces authentication at edge nodes globally
- GitHub Actions runs daily: mounts S3 folder, runs automation, syncs results back, invalidates CloudFront cache
Access Control: Basic HTTP authentication (username/password) enforced at CloudFront. No backend needed. Users authenticate once in their browser, then browse the genome naturally.
Why This Approach Works
We didn’t have a development team or a large budget. So we focused on composition: choosing tools that solve 80% of the problem, automating the 20% they don’t handle.
This approach has three advantages:
It scales. As new collaborators join and new data arrives, the same automation handles it. No new code needed. The GitHub Action handles everything.
It’s maintainable. The GitHub Action runs on a schedule you set. The conversion script is pure Python—transparent and understandable. Version control means you can roll back if something breaks.
It’s focused on the science. Researchers spend time analyzing genes, not wrestling with deployment pipelines. They add a CSV file to a folder. Within minutes, it’s browsable alongside all their collaborators’ data.
The Lesson
You don’t need a development team to build research infrastructure. You need:
- Clear problem definition (one gene, all experiments)
- Existing tools that solve most of it (IGV for browsing)
- Automation to handle the gaps (Python script + GitHub Actions)
If you have a similar challenge—multiple collaborators, heterogeneous data, limited resources—this approach works. Look for the existing tool that’s 80% of your solution. Build automation for the 20% it doesn’t cover. Deploy it once, then let it run.
The full code is available on GitHub. It includes:
- The Python automation pipeline
- AWS deployment scripts
- GitHub Actions workflow
- Complete documentation for adding new datasets
Limitations and Future Work
- Cross-species support: Currently handles mouse→human translation for RNA-seq only. Other species would need custom ortholog mappings. Other data types need additional code.
- User management: Today uses a shared password. For multiple independent users, consider Amazon Cognito or similar.
These aren’t blockers for most research groups, but they’re worth noting if you’re adapting this approach.